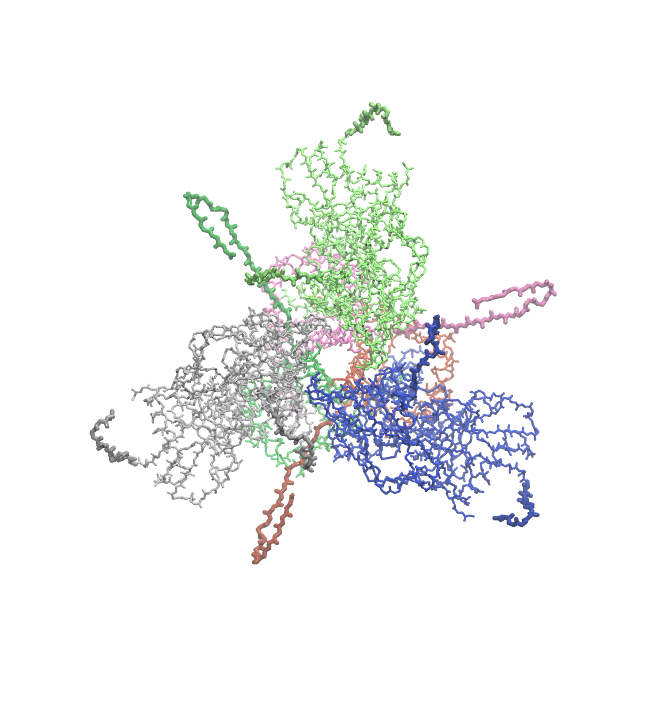
Example 7: Closed, Unliganded HIV-1 BG505 Env SOSIP.664 Trimer¶
PDB ID 4zmj is a structure of the HIV-1 Env BG505 SOSIP-664 ectodomain trimer in a closed conformation, without any ligands bound. It is one of the earliest X-ray crystal structures solved for this protein in trimeric form.
Pestifer understands how to build a system using any chosen biomolecular assembly available in the structure file. In the case of 4zmj, the asymmetric unit is a single heterodimeric protomer composed of chains G (gp120) and B (gp41). The relevant biological assembly is a C3-symmetric homotrimer of protomers, which is labeled biological assembly 1 in the PDB header. Here we specify that the new chains generated by BIOMT transforms are H and J for chain G and C and D for chain B. Pestifer will also by default undo any engineered mutations (there are three in 4zmj) and add any unresolved or zero-occupancy residues. A build of the 4zmj trimer illustrates these capabilities.
# Author: Cameron F. Abrams, <cfa22@drexel.edu>
#
# pestifer input script
#
# Simple build of solvated HIV-1 Env SOSIP ectodomain trimer 4zmj
#
# Notes:
# - Chain G produces daughter chains H and J upon BIOMT transformation to build trimer
# - Chain B produces daughter chains C and D upon BIOMT transformation to build trimer
# - Existing chains C and D (these are glycans) are assigned new chainIDs
# - All glycans are assigned chainIDs that are not shared with any protein
# - Missing protein is built-in
# - Fine-tuning ramachandran rotations performed at N-terminus of HN1
# to avoid steric clashes when building in these missing residues
# - A five-phase NPT equilibration is used to settle the density
# - A production tarball is generated: prod_4zmj.tgz
#
title: Closed, Unliganded HIV-1 BG505 Env SOSIP-664 Trimer
tasks:
- fetch:
sourceID: 4zmj
- psfgen:
source:
biological_assembly: 1
transform_reserves:
G: [H,J]
B: [C,D]
mods:
mutations: # undo the SOSIP mutations
- G:CYS,501,ALA
- B:PRO,559,ILE
- B:CYS,605,THR
crotations:
- psi,B,546,568,-180.0
- phi,B,547,568,-60.0
- validate:
tests:
- attribute_test:
name: point mutation
selection: protein and chain G H J and resid 501 and name CA
attribute: resname
value: ALA
value_count: 3
- attribute_test:
name: point mutation
selection: protein and chain B C D and resid 559 and name CA
attribute: resname
value: ILE
value_count: 3
- attribute_test:
name: point mutation
selection: protein and chain B C D and resid 605 and name CA
attribute: resname
value: THR
value_count: 3
- md:
ensemble: minimize
- ligate:
steer:
nsteps: 4200
- validate:
tests:
- connection_test:
name: gp120 gaps ligation
selection: protein and chain G H J and ((resid 185 and insertion I) or resid 187 410 411)
connection_type: interresidue
connection_count: 6
- connection_test:
name: gp41 gaps ligation
selection: protein and chain B C D and resid 568 569
connection_type: interresidue
connection_count: 3
- md:
ensemble: minimize
- md:
cpu-override: True
ensemble: NVT
nsteps: 2400
- solvate:
- md:
ensemble: minimize
- md:
ensemble: NVT
- md:
ensemble: NPT
nsteps: 200
- md:
ensemble: NPT
nsteps: 400
- md:
ensemble: NPT
nsteps: 800
- md:
ensemble: NPT
nsteps: 1600
- md:
ensemble: NPT
nsteps: 13200
- mdplot:
timeseries:
- density
- - a_x
- b_y
- c_z
grid: True
basename: solvated
- terminate:
basename: my_4zmj
artifacts: artifacts
package:
basename: prod_4zmj
namd:
ensemble: NPT
Step |
Task |
Details |
|---|---|---|
1 |
|
|
2 |
|
mutations, crotations |
3 |
|
3 test(s) |
4 |
|
minimize |
5 |
|
ligate chain breaks (steered MD, 4,200 steps) |
6 |
|
2 test(s) |
7 |
|
minimize → NVT (2,400 steps) |
8 |
|
water box |
9 |
|
minimize → NVT (2,000 steps) → NPT (16,200 steps, 5 phases) |
10 |
|
equilibration time-series plots → |
11 |
|
basename: |
There are several new aspects in this example relative to the first four. First, in the psfgen task, the source directive has a biological_assembly specification with transform_reserves and sequence subdirectives.
Clearly we are indicating biological assembly 1, which you can verify through the RCSB web interface or by reading the PDB file header is the trimer.
There is also a ligate task. Together, the loops subdirective of the sequence directive in the source, and the ligate task, constitute the method of inserting missing residues (residues designated by MISSING records in the PDB or zero-occupancy in the mmCIF). Building in missing protein loops that are internal to any given chain is done in the following way:
Via
residuecommands insidesegmentstanzas of thepsfgenscript, each missing residue is indicated. Additionally, a sacrificial glycine residue is added at the C-terminus of any run of missing residues. After allsegmentsare defined, sacrificial glycines are deleted and NTER/CTER patches are explicitly added. Whenpsfgenis run, theguesscoordscommand results in this missing residues being built in according to their default internal coordinates; this means they grow in as straight chains where every phi and psi angle is 180 degrees.Each such loop is put through a
declashingprocedure in which phi and psi angles are adjusteds so that the loop residues do not clash with any other residues. Sometimes, additional manual adjustment of some dihedrals is necessary; that is the case here.After a minimization, pestifer then runs a
ligationtask which has two parts. First, a steered MD is run that shrinks the distance between the terminal glycine of each loop and the location on the resolved protein to which it should be attached. Second, anotherpsfgenrun is performed to “heal” the gap between the C-terminus of the loop and its attachment point with a peptide bond.
Declashing is done using a Monte-Carlo approach where trial rotations are suggested and only performed if they result in a reduction in the number of steric clashes.
The 4zmj entry contains partially resolved glycans. By default, pestifer will include all resolved glycans. These can be excluded using an excludes list that specifies resnames like NAG, MAN, etc.
The snapshots below illustrate the process by which the loops are grown in. In these snapshots, only backbone protein atoms are shown with bonds drawn as lines. The model-built parts are drawn with thick bonds, and the six chains are colored uniquely.